Lab 1
Lab 1-Recrystallization Lab
|
Principal IR Absorptions for Certain Functional Groups |
||
|---|---|---|
|
Functional Group Names & Example compounds |
Absorption Ranges(cm-1) [Look for a single absorption in these regions, unless stated otherwise.] |
Type of Vibration causing IR absorption
|
|
Alkanes:
|
3000-2800 (Note: The absorptions can be seen as several distinct peaks in this region.) |
H-C-H Asymmetric & Symmetric Stretch
|
|
1500-1440 |
H-C-H Bend |
|
|
Alkenes:
|
3100-3000 |
C=C-H Asymmetric Stretch |
|
1675-1600 |
C-C=C Symmetric Stretch |
|
|
Alkynes:
|
3300-3200 |
|
|
2200-2100 |
|
|
|
Aromatic Rings:
|
3100-3000 |
C=C-H Asymmetric Stretch |
|
1600-1580 |
C-C=C Symmetric Stretch |
|
|
1500-1450 |
C-C=C Asymmetric Stretch |
|
|
Phenols & Alcohols:
|
3600-3100
(Note: Phenols MUST have Aromatic Ring Absorptions too.)
|
Hydrogen-bonded O-H Stretch
(This peak usually appears much broader than the other IR absorptions.
|
|
Carboxylic Acids:
|
3400-2400 (This peak always covers the entire region with a VERY BROAD peak.)
|
Hydrogen-bonded O-H Stretch [Note: This peak can obscure other peaks in this region.] |
|
1730-1650 |
C=O Stretch |
|
|
Ketones:
|
1750-1625 |
C=O Stretch |
|
Aldehydes:
|
1750-1625 |
C=O Stretch |
|
2850-2800 |
C-H Stretch off C=O |
|
|
2750-2700 |
C-H Stretch off C=O |
|
|
Esters:
|
1755-1650 |
C=O Stretch |
|
(1300-1000) |
(C-O Stretch) |
|
|
Ethers:
|
(1300-1000) |
(C-O Stretch) |
|
Amines—Primary:
|
3500-3100 (TWO PEAKS!) |
N-H Stretch |
|
1640-1560 |
N-H Bend |
|
|
Amines—Secondary:
(Note: Tertiary amines have no N-H absorptions. Therefore, you won’t see any evidence of a tertiary amine/amide in your FTIR.) |
3500-3100 (ONE PEAK!) |
N-H Stretch |
|
1550-1450 |
N-H Bend |
|
|
Nitriles:
|
2300-2200 |
|
|
Nitro Groups:
(Note: Both peaks are <200 cm-1 apart.)
|
1600-1500
|
N=O Stretch
|
|
1400-1300 |
N=O Bend |
|
|
Amides:
|
3500-3100 |
N-H Stretch (similar to amines) |
|
1670-1600 |
C=O Stretch |
|
|
1640-1550 |
N-H Bend |
|















An interactive web tutorial on Infrared Spectroscopy and Organic Functional Groups is listed below. It provides more specific information about how you can use IR spectroscopy to identify functional groups. http://www.wellesley.edu/Chemistry/Flick/irmain.html
Lab 1
Recrystallization
Pre-lab Work:
1. Reading Assignment
· Technique 1 - Safety: Section 1.
· Technique 3 – Notebook/Percent Yield: Section 3.
· Technique 4 - Glassware: Sections 4.2 & 4.4.
· Technique 15 - Recrystallization: Sections 15.1-15.5 & 15.8.
· Gas Chromatography-Mass Spectroscopy - Chem. 211 Online Lab Manual Appendix.
· Description of Experiment (below)
Introduction:
It is frequently necessary to purify an organic compound before an accurate analysis of the material can be achieved. Organic solids can often be purified by recrystallization. Compounds having different solubilities at different temperatures can generally be recrystallized; in practice this means that the solid is soluble ONLY in hot solvents and insoluble otherwise. The art of recrystallization is best learned from experience using well-behaved compounds and solvents or solvent mixtures; purity of the separated material using gas chromatography can be used to check your technique. With unknown compounds a number of solvents are generally tried before choosing one giving the best results. The purity and identity of your recrystallized unknown can be determined if provided a list of possible compounds (see below) and your recrystallized unknown’s gc/ms data.
Before coming to the laboratory, review the steps in recrystallization summarized in sec. 15.5 of Mohrig. Since the operations will be performed on a small scale, the time factor for each recrystallization is relatively short. With practice you should be able to do a small-scale recrystallization in only a few minutes.
The following is a list of some well-behaved sample compounds with their respective melting points and solvents that may be useful for their recrystallization. These representative solvents range from non-polar (hexanes) to intermediate polarity (ethanol) and finally to highly polar (water).
|
acetanilide (mp = 115oC)
|
benzoic acid (mp = 122-123oC)
|
2-methylbenzoic acid (mp = 103-105oC)
|
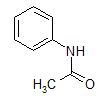
Solvents available: [see Table 15.1 of Mohrig for boiling points]
Hexanes, ethanol, water
Given one of the compounds above with an approximately 10% impurity in it (as seen in the GC spectrum below for one of the impure unknowns), determine its solubility, select a suitable solvent(s) for recrystallization, and recrystallize the material. You will determine the purity of the recrystallized unknown by gas chromatography and identify your unknown from its mass spectrum.
|
Fig. 1: Gas Chromatograph of one of the unknowns with 10% impurity (at 3.9 minutes) |

Safety:

Experimental:
Note: ALL boiling solutions (in beakers, flasks, or test tubes) should contain a boiling stick/glass rod. (Why?)
Determination of Solubility
Place approximately 50 mg of solid (about the amount that will fit on the tip of a spatula) into a small test tube. Add approximately 0.5 mL of solvent at room temperature. Record whether or not the solid dissolves at room temperature. If the solid does not dissolve, heat the test tube in a beaker of boiling recrystallization solvent, adding up to 1.0 mL more of boiling solvent drop-wise until the solid just dissolves. (Why do you place the test tube in the beaker containing the same solvent?) Again, record whether or not the solid dissolves in boiling solvent. Test the solubility of your compound with hexanes, with ethanol, and with water.
Selection of Recrystallization Solvent
A solvent in which a solid dissolves at room temperature is not a suitable solvent for single-solvent recrystallization of the material. (Why?) Consider which solvents each of your samples did not dissolve in at room temperature yet dissolved completely when heated. Take these sample tubes (from determination of solubility) and chill in an ice bath to determine if recrystallization will occur. Create a table for your observations and conclusions based on your results. If none of the single-solvent systems were effective, than see your instructor about using hot filtration or a mixed-solvent system to purify your unknown.
Recrystallization using a single solvent:
Heat the chosen solvent to its boiling point in a beaker. Use a steam bath or hot plate depending on the boiling point of the solvent. (Why shouldn’t you use a steam bath to boil water?). Place 0.2 to 0.3g of solid in a small Erlenmeyer flask and place the flask on the heat source next to the beaker of boiling solvent. Immediately add boiling solvent to barely cover the solid. STIR the mixture to increase the solubility of the solid. Maintain the flask at the bp of the solvent, continuing to add increments of boiling solvent until the solid dissolves. Set the flask on the lab bench and let the contents cool slowly, undisturbed, to room temperature. Chill the flask in an ice bath. Collect the crystals by vacuum filtration (See Fig. 10.6 in Mohrig-no ‘trap’ will be used). Use a small Hirsch funnel, instead of a Buchner funnel (Why?), and rinse the crystals with a few mL of ice-cold recrystallization solvent. (Why cold solvent?) Dry the crystals by spreading them on a porcelain tile square.
Prepare a gc/ms sample using 0.05-0.1 grams of your dry, recrystallized solid in a small test tube with approximately 1-1.5 mL of methanol. Make sure your solid dissolves completely before filling your gc/ms vial more than ½ full with your solution.
Waste Disposal:
Once you have identified your unknown, dispose of it in the flammable waste bottle rinsing any glassware with acetone.
Results:
- Create a data table to display the following information: Unknown number, recrystallization solvent, and the structure and name of your unknown.
- Justify the identity of your unknown using the MS data of the most abundant GC peak. Use a minimum of two masses to confirm your unknown.
- Using the “percent report” of your gc/ms, determine the relative purity of your unknown. The impurity used in your sample appears at approximately 3.9 minutes in your GC.
Questions to ponder…
1. How does recrystallization purify an impure compound?
2. Discuss how melting points of the impure sample and recrystallized sample could have been used to determine the success of your recrystallization and identity of your unknown.
3. Discuss any influences/errors in your experiment and the magnitude of these influences/errors on your final results. What would you do differently if you were to do this lab again?
(Please read the section on “Experimental Errors” given at the beginning of this lab manual. Note, ‘influences’ are not errors, but are typically procedural steps that had a significant impact on your final results.)

